凌恩生物文献分享|大热门--HiFi宏基因组有效提升真核生物群落检出效果

binning和HiFi宏基因组测序优势:
不依赖于微生物的分离培养,环境微生物单菌基因组(框架图)研究的一种新的途径和高性价比策略;
可以得到环境中丰度较低的宏基因组,为研究低丰度微生物提供了途径;
引入了宏观生态的研究理念,对环境中微生物菌群的多样性、功能活性等宏观特征进行研究,可以更准确地反应出微生物生存的真实状态。

美国国家海洋和大气管理局近期在《mSystems》期刊上(IF=7.324)发表的“Long-Read Sequencing Improves Recovery of Picoeukaryotic Genomes and Zooplankton Marker Genes from Marine Metagenomes”研究论文中,验证了HiFi宏基因组binning的结果可以改善海洋群落组成分析,并为真核生物植物和浮游动物的遗传学提供重要的见解。

期刊:mSystems
影响因子:7.324
发表时间:2022
样本类型:水样
一、实验背景
三代宏基因组测序具有改善宏基因组组装的潜力,并提供了比二代测序更稳健的微生物群落组成和功能的评估。高通量测序可对无法培养分类群为主的生态重要生境进行群落调查,通过组装基因组 (MAG) 对原核生物多样性和生态学提供了重要的见解。与原核生物相比,真核生物基因组的binning相关计算更大,更大的基因组、内含子的存在和染色体间的组成差异,是真核 MAGs 准确矫正的重要障碍。
二、实验设计
将PacBio CCS HiFi 测序应用于14个海洋水柱样本,并将结果与相应环境 DNA 样本的二代宏基因组的结果进行比较。
三、实验结果
1、二代和三代测序结果与组装
每个样本的PacBio CCS 测定reads 的数量为6,807~1,907,840个,相应的 Illumina reads数量为2,879,847~264,220,592。
文章对相应eDNA样品的不同组装方式进行了比较,Illumina组装、PacBio(HiFiasm meta和metaFlye)以及Illumina和PacBio的混合组装(hybridSPAdes)。经比较,相比于二代或混合组装中的contigs,仅三代产生的组装中的最长contigs更长。最长contigs(> 800 kb,来自Las19c139_27m-3)上的ORF注释为Verrucomicrobiaceae 细菌TMED86(图1)。
其他组装方式显示contigs之间的差异很大;混合组装和二代比三代可以组装更多的 > 1kb的contigs, 另外metaFlye 组装出来contigs数量 (> 5kb)是最多的。
2、二代和三代测序注释比较
使用隐马尔可夫模型 (HMMs) 比较了从Illumina、PacBio和混合组装中提取的小亚基 SSU rRNA基因,混合和二代组装之间 SSU rRNA 序列的平均数量相当。基于 K-mer ,三代和二代样本之间的物种注释组成比较差异很大。三代和二代宏基因组的基因组的多样性差异很大(图1)。
α多样性分析显示 Illumina 和混合组装的宏基因组的数值范围比 PacBio 宏基因组要小很多,Shannon 多样性值和基于组装分析差异很大(图2)。基于reads以及组装的注释分类学比较,β多样性显示三代和二代样本之间差异有统计学意义,混合组装与 Illumina metaSPAdes 组装紧密聚集。
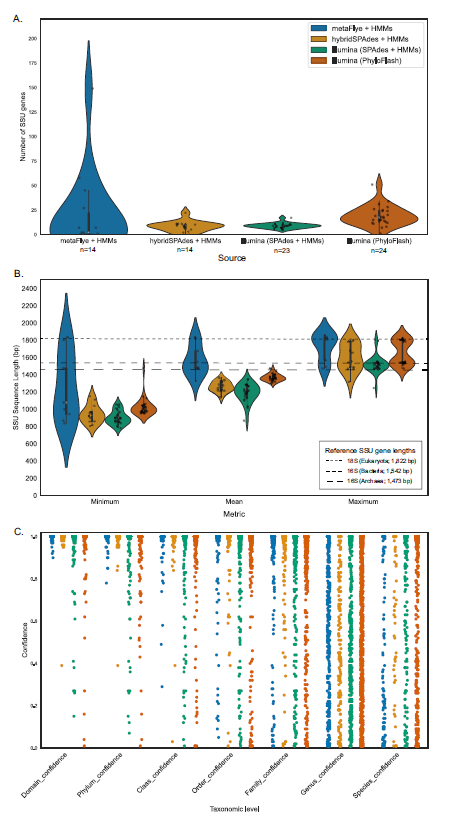
图1以四种不同方式组装的 16S 和 18S rRNA 基因
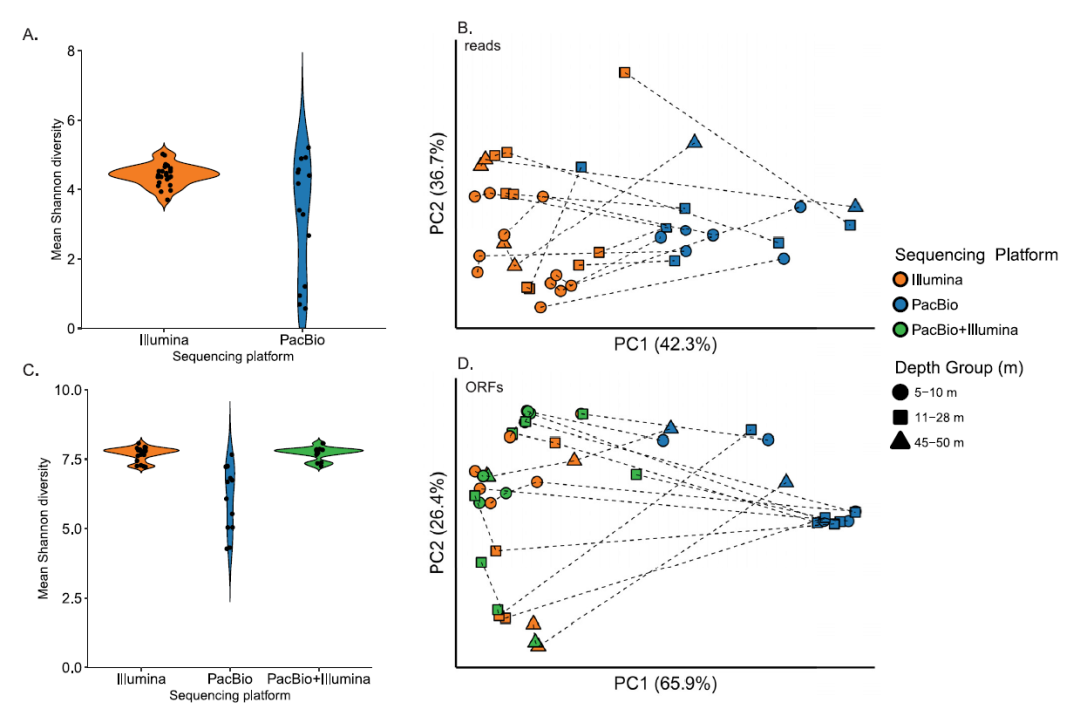
图 2 未组装的二代和三代和从三代、二代和混合组装中提取的开放阅读框 (ORF) 的α和β多样性分析
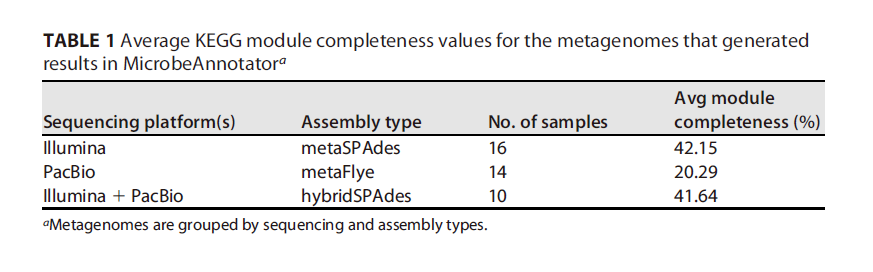
三代测序的主要潜在优势之一是矫正完整的蛋白编码基因和重建完整的生化基因通路。因此文章比较了所有组装类型中 ORF 的总数、具有 KEGG 注释的 ORF 的分数和每个contigs注释 ORF 的平均数量(图3)以及比较了完整(> 80%)KEGG模块的数量(表1),发现二代的平均模块完整性最高为42%,而三代的平均值不到该值的一半。二代和混合组装体的总 ORF 数量最高(图3A),但三代组装得到的MAG具有更高比例的注释ORF(图3B),从而导致所有组装类型中总注释 ORF 的数量相当(图3C)。
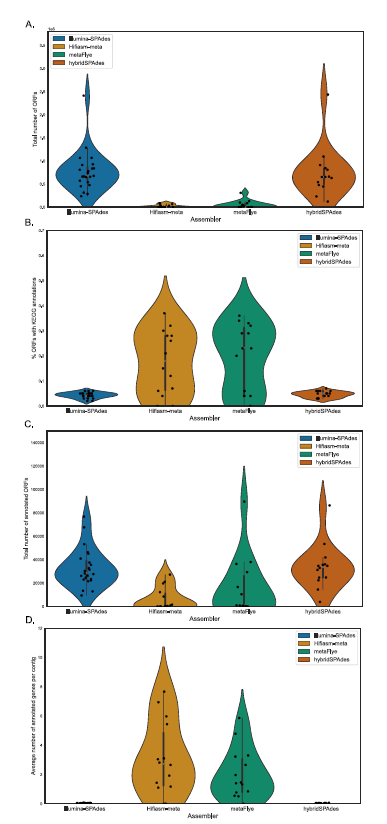
图3 四种组装类型中提取的带注释和不带注释的开放阅读框 (ORF)
表 1 MicrobeAnnotator 结果中平均 KEGG 模块完整性值
3、二代和三代测序binning比较
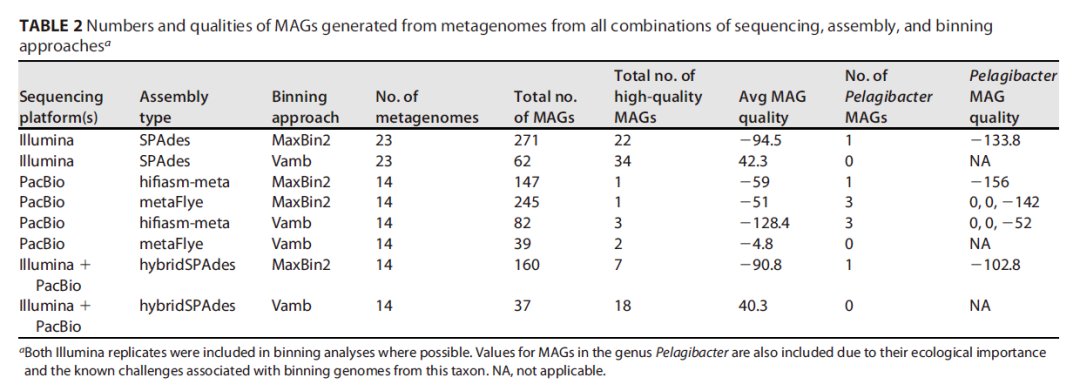
使用两种不同的binning程序比较了二代、三代和混合组装生成的 MAGs 的数量和质量评分。总体而言,使用 Vamb binning的混合组装产生了最多的MAG(平均值为18)以及40.3的高质量评分(表2)。
表2测序、组装和binning方法所有组合的宏基因组生成的 MAGs 的数量和质量
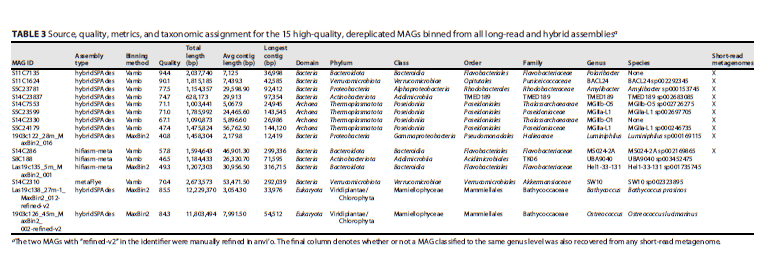
在所有三代和混合组装产生的15个高质量的 MAGs 中,从相应二代宏基因组中匹配到的MAG缺少(在属水平上定义)5个(表3)。这些独特的 MAGs 中包括疣微菌门SW10、酸杆菌门TK06 和黄杆菌科微生物。其中两个 MAGs 可能受益于 PacBio 组装产生的较长contigs,最长的 MAG contigs在290,000和320,000 bp之间(表3)。同样从二代宏基因组中复原的MAG中,10个中有9个来自混合组装,其中8个来自Vamb binning程序。
表3来自所有三代和混合组装的15个高质量MAG的来源、质量和分类结果
此外,从混合组装中产生了两个真核MAGs。经过人工优化(图4)的单拷贝基因的分类和DNA依赖性的rRNA聚合酶基因的系统发育定位,它们被鉴定为微真核生物类群——Ostreococcus lucimarinus 和Bathycoccus prasinos。
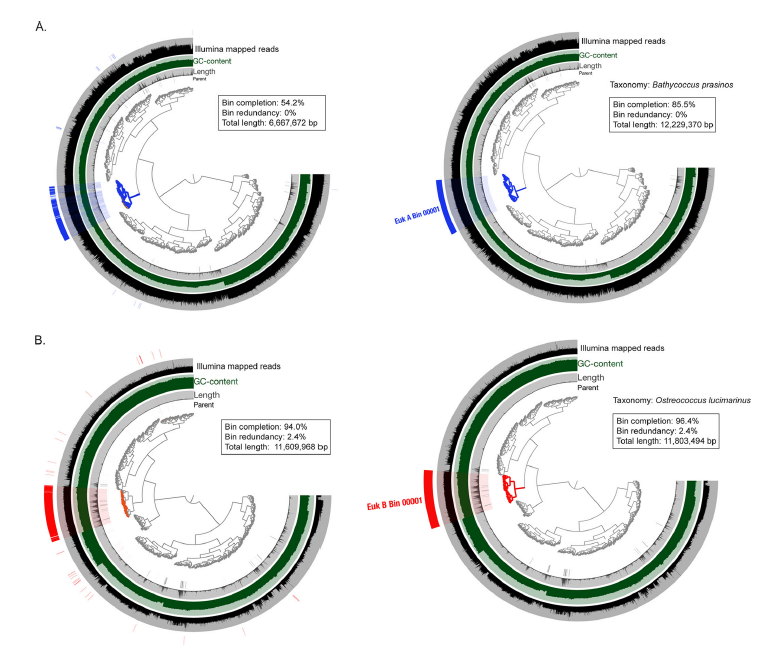
图4 人工重组前后微真核 MAGs 及其来源宏基因组的可视化表示
4、真核 rRNA 基因序列
来自5个不同 metaFlye 三代组装的9个 > 10000 bp的contigs包含全长 18S rRNA 和完整或部分 28S rRNA 基因序列。根据位于contigs上的 18S 和 28S rRNA 基因序列,进行浮游动物物种注释。在9个 18S rRNA 基因中,2个对磷虾种 Euphausia pacifica ,3个对桡足类种 Calanus pacificus 或 Metridia sp. 的 BLAST 相似度最高。。
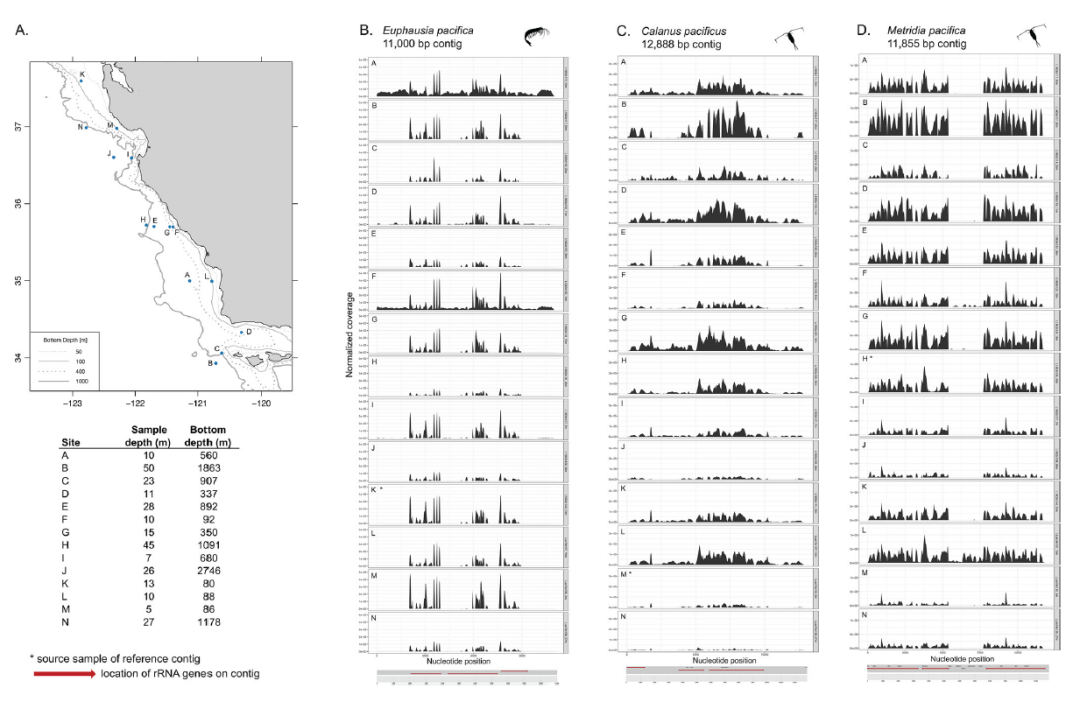
图5三代组装产生的三个contigs的二代宏基因组覆盖度
四、研究结论
与单独的扩增子测序或二代宏基因组测序相比,二代+三代宏基因组的组合可以得到更完整的浮游群落组成结构,包括磷虾和桡足类。这项研究为理解物种间遗传变异性和利用 eDNA 序列数据补充海洋生态系统中物种调查结果提供了基础。
凌恩生物优推PacBio HiFi三代扩增子、宏基因组测序项目,为您提供专业优质的三代全长16S、ITS测序,各类环境、医学样本宏基因组测序及专业可靠的分析服务。测序找凌恩,组学科研好伙伴~
参考文献
Patin NV,Goodwin KD. Long-Read Sequencing Improves Recovery of Picoeukaryotic Genomes and Zooplankton Marker Genes from Marine Metagenomes. mSystems. 2022;7 (6):e0059522. doi:10.1128/msystems.00595-22